Chapitre 8 Cellule cancéreuse et tissu cancéreux
 Objectifs
Objectifs Décrire les bases moléculaires de la cancérogenèse et connaître quelques exemples dans chacune des trois grandes familles de gènes (oncogènes, gènes suppresseurs et gènes de l’homéostasie génétique).
Décrire les bases moléculaires de la cancérogenèse et connaître quelques exemples dans chacune des trois grandes familles de gènes (oncogènes, gènes suppresseurs et gènes de l’homéostasie génétique).
Connaître les grands mécanismes de régulation de l’expression ou de la fonction de ces gènes.
Connaître les principaux facteurs de risque génétiques et environnementaux des cancers.
Connaître des exemples de dérégulation du cycle cellulaire et de l’apoptose dans les cancers.
Décrire les caractéristiques biologiques et morphologiques d’une cellule cancéreuse. Décrire les cellules constituant généralement le stroma des tumeurs.
Connaître les principales caractéristiques de la vascularisation des tumeurs
Connaître les grands mécanismes de réponse immune anti-tumorale.
La maladie cancéreuse se caractérise par l’envahissement progressif de l’organe d’origine, puis de l’organisme entier, par des cellules devenues peu sensibles ou insensibles aux mécanismes d’homéostasie tissulaire et ayant acquis une capacité de prolifération indéfinie (immortalisation).
Ces cellules tumorales dérivent dans la grande majorité des cas d’une seule cellule (monoclonale). Les particularités des cellules tumorales sont liées à l’accumulation d’altérations de leur génome (génotype). Ces altérations sont le plus souvent acquises au cours de la genèse tumorale, mais certaines peuvent être d’origine héréditaire (prédispositions familiales).
Les clones tumoraux peuvent perdre ou conserver certaines caractéristiques morphologiques et fonctionnelles des cellules originelles, ou en acquérir de nouvelles (variabilité du phénotype des sous-clones).
Ces modifications vont s’inscrire à la fois dans le noyau, dans le cytoplasme et sur la membrane des cellules pathologiques.
Bases moléculaires du cancer
Un néoplasme est la conséquence d’altérations successives du génome des cellules tumorales, qui perturbent de façon permanente l’homéostasie tissulaire (figure 8.1).
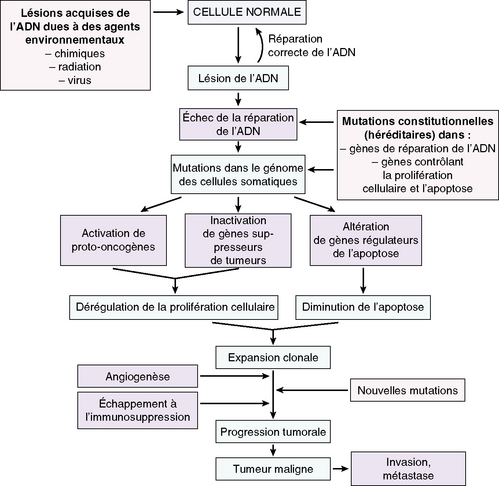
Dans la cellule cancéreuse, il y a rupture permanente de l’équilibre entre les signaux intracellulaires :
La coexistence de plusieurs événements est nécessaire à la transformation cancéreuse. L’activation de nouveaux oncogènes se poursuit tout au long de la progression tumorale : processus multi-étapes.
Différents agents de l’environnement conduisent au développement d’un cancer
• Agents initiateurs : ils induisent une lésion définitive de l’ADN (ex : mutation, cassure). Souvent, ces carcinogènes sont activés par des réactions métaboliques.
• Agents promoteurs : ils favorisent l’expression d’une lésion génétique, préalablement induite par un agent initiateur. Ils n’induisent pas de lésions de l’ADN. Le temps écoulé entre l’initiation et l’apparition des tumeurs est réduit en présence d’agents promoteurs.
Les trois familles de gènes impliquées dans la cancérogenèse
Oncogènes
Certains virus animaux sont capables d’induire des tumeurs (ex : sarcome de Rous du poulet, découvert en 1911). Les propriétés transformantes de ces virus sont dues à la présence dans leur génome de séquences particulières, les oncogènes viraux (v-onc).
Ces gènes renferment à eux seuls toute l’information pour l’activité transformante. Ces gènes sont des formes altérées de gènes normaux d’origine cellulaire, les proto-oncogènes, capturés par les rétrovirus au cours de leur réplication.
Les proto-oncogènes sont conservés dans toutes les espèces (de l’insecte à l’homme) et jouent un rôle essentiel dans des étapes clés de la régulation de l’embryogénèse ou de la croissance cellulaire ou tissulaire. Ces gènes normaux lorsqu’ils sont remaniés et/ou sur-exprimés deviennent des oncogènes (c-onc). Ils peuvent induire l’apparition et/ou le développement d’une tumeur.
Les oncogènes sont schématiquement classés en :
• gènes immortalisants (ex : c-myc) codant pour des protéines nucléaires se liant à l’ADN ;
• gènes transformants (ex : KRAS, RET, KIT) (tableau 8.1).
Tableau 8.1 Exemples de proto-oncogènes impliqués dans des tumeurs humaines.
| Proto-oncogènes | Type d’anomalie | Exemples de tumeurs impliquées |
|---|---|---|
| ERBB1 (EGFR) | sur-expression ou mutation activatrice | nombreux carcinomes |
| ERBB2 (HER2) | amplification | carcinomes mammaires et ovariens |
| FLT3 | mutation activatrice | leucémies aiguës myéloïdes |
| RET | mutation activatrice | carcinomes thyroïdiens |
| PDGFR | mutation activatrice | sarcomes, gliomes |
| KIT | mutation activatrice | tumeurs stromales gastro-intestinales |
| KRAS | mutation activatrice | carcinomes coliques, bronchiques, pancréatiques |
| NRAS | mutation activatrice | leucémies, mélanomes |
| BRAF | mutation activatrice | mélanomes |
| ABL | translocation | leucémie myéloïde chronique |
| CMYC | translocation | lymphome de Burkitt |
| NMYC | amplification | neuroblastomes |
| cycline D | translocation | lymphomes du manteau |
| CDK4 | mutation activatrice | mélanomes |
Gènes suppresseurs
Les gènes suppresseurs de tumeur (ou anti-oncogènes) sont des inhibiteurs de la croissance cellulaire. L’inactivation du produit de ces gènes par perte de fonction biallélique se traduit par l’absence d’un signal de non-prolifération cellulaire : il s’agit d’une perte de fonction.
Le premier gène suppresseur de tumeur décrit est le gène Rb du rétinoblastome. Le gène suppresseur de tumeur le plus souvent impliqué est la TP53, avec des mutations somatiques dans de très nombreux cancers et des mutations germinales dans le syndrome de Li-Fraumeni.
Les oncogènes et gènes suppresseurs de tumeur codent pour des protéines qui interviennent dans les grandes fonctions cellulaires : signalisation, prolifération, différenciation, cycle, apoptose (tableau 8.2).
Tableau 8.2 Exemples de gènes suppresseurs de tumeurs impliqués dans des tumeurs humaines.
| Gènes suppresseurs | Exemples de tumeurs impliquées |
|---|---|
| TP53 | nombreux cancers |
| NF1 | tumeurs des nerfs périphériques |
| NF2 | méningiomes |
| APC | carcinomes digestifs |
| WT1 | néphroblastome (tumeur de Wilms) |
Gènes de maintien de l’intégrité (care takers)
Des agents pathogènes (rayons X, UV, hydrocarbures) peuvent entraîner des lésions ponctuelles de l’ADN (cassure d’un brin, délétion, mutation d’une base). Les gènes de maintien de l’intégrité codent pour un complexe multi-fonctionnel capable de surveiller l’intégrité du génome (MSH2, MSH6.). En cas d’anomalies, différents systèmes de réparation sont mis en place (BRCA1, rad50, MLH-1). S’ils échouent, la cellule lésée meurt par apoptose.
L’altération des 2 allèles de ces gènes conduit à une susceptibilité accrue aux cancers, par instabilité génétique (accumulation de mutations conduisant à l’activation d’oncogènes ou à l’inactivation d’anti-oncogènes).
Des mutations impliquant ces trois familles de gènes sont présentes dans la majorité des cancers. Ces lésions peuvent être d’origine environnementale, sous l’effet notamment d’agents initiateurs, ou au contraire d’origine génétique.
Contrôle de l’expression et/ou de l’activation
Des proto-oncogènes, des gènes suppresseurs de tumeurs et des gènes du maintien de l’intégrité du génome.
Plusieurs mécanismes peuvent être responsables de l’expression et/ou de l’activation des gènes impliqués dans la tumorigenèse. Ces mécanismes ne sont pas mutuellement exclusifs.
Mutations ponctuelles, délétions, insertions (figure 8.2)
Pour les proto-oncogènes, un seul événement génétique est généralement suffisant pour l’activation (dominant). Pour les gènes suppresseurs de tumeurs et les gènes de surveillance du génome, un double événement est nécessaire pour que le gène soit inactivé au niveau des 2 allèles (récessif).
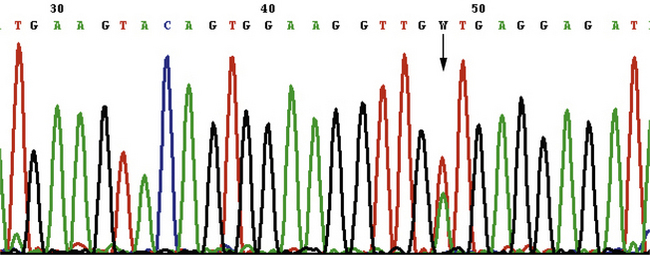
Figure 8.2 Mutation « gain de fonction » du proto-oncogène KIT dans une tumeur stromale digestive. Le séquençage après amplification de l’ADN extrait des cellules tumorales permet de mettre en évidence une substitution d’une base (en position 48 sur ce graphe), correspondant à la mutation c.1679 T > A p.V560D. Cette délétion est responsable de l’expression d’une protéine oncogénique, car constitutivement activée.
Amplification génique
Ce phénomène correspond à une multiplication du nombre de copies d’un gène. Il en résulte une augmentation de son expression. Il serait surtout tardif dans l’oncogenèse (figure 8.3).
Réarrangements chromosomiques
Les translocations peuvent aboutir soit à l’expression d’une protéine chimérique résultant de la fusion entre deux gènes, soit à l’hyperexpression d’un oncogène en raison de la transposition de la région codante de celui-ci à proximité de séquences régulatrices d’autres gènes.
Exemple 1 : Dans la leucémie myéloïde chronique (LMC) la translocation réciproque entre les chromosomes 9 et 22 produit un chromosome 22 raccourci : le chromosome de Philadelphie. Cette translocation aboutit à un gène de fusion bcr/c-abl codant pour une tyrosine kinase activée. Il existe actuellement une molécule thérapeutique capable de bloquer spécifiquement la tyrosine kinase activée par cette translocation. Grâce à cette thérapeutique (Imatinib), le pronostic de la LMC a été transformé.
Exemple 2 : Dans le lymphome de Burkitt, la translocation (8; 14) aboutit à la surexpression de l’oncogène c-myc (chromosome 8) qui se retrouve sous le contrôle du promoteur de la chaîne lourde des immunoglobulines (chromosome 14).
Facteurs favorisant les altérations
Des proto-oncogènes, des gènes suppresseurs de tumeurs et des gènes du maintien de l’intégrité du génome.
Facteurs héréditaires
Ces facteurs génétiques sont responsables de prédispositions familiales aux cancers. La transmision peut être dominante ou récessive, et la pénétrance variable. Les prédispositions génétiques aux cancers sont nombreuses, et les prédispositions monogéniques sont les mieux connues (tableau 8.3).
Tableau 8.3 Exemples de prédispositions familiales aux tumeurs chez l’homme.
| Gènes | Tumeurs ou prédisposition familiale |
|---|---|
| Proto-oncogènes | |
| MEN1, RET KIT, PDGFRA CDK4 |
néoplasies endocriniennes multiples tumeurs stromales gastro-intestinales mélanomes |
| Gènes suppresseurs de tumeur | |
| RB TP53 NF1 |
rétinoblastome syndrome de Li Fraumeni tumeurs nerveuses (neurofibromatose) |
| Gènes impliqués dans le maintien de l’intégrité de l’ADN | |
| XPA BRCA1 MLH1, MSH2 |
xeroderma pigmentosum de type A, tumeurs cutanées carcinomes mammaires et ovariens carcinomes colorectaux |
Facteurs viraux
• Rétrovirus à ARN. Certains rétrovirus sont directement oncogéniques, mais il n’en existe d’exemple connu que chez l’animal. Chez l’homme, le rétrovirus HTLV1 s’intègre au hasard dans le génome, il est dépourvu d’oncogène mais contient un gène transactivateur (tax) capable d’activer les gènes de l’interleukine 2 et de son récepteur dans les lymphocytes T.
• Virus oncogènes à ADN : ils ne renferment pas d’oncogène de type v-onc. Le plus souvent ils semblent agir par trans-activation de gènes cellulaires (mutagénèse insertionnelle).
Exemple : Le virus d’Epstein-Barr induit chez les sujets immunodéprimés (VIH, endémie paludique, transplantés) une intense prolifération polyclonale des lymphocytes B infectés et augmente ainsi le risque de survenue de translocations chromosomiques. Au cours de ces translocations somatiques peuvent se produire des juxtapositions accidentelles de gènes, capables d’activer des proto-oncogènes : la translocation t(8; 14) : juxtaposition de c-myc et du gène de la région constante des immuno-globulines.
Progression tumorale et cycle cellulaire
La progression du cycle cellulaire est finement régulée par des « points de contrôle », qui permettent notamment une régulation de la vitesse de prolifération et un maintien de l’intégrité du génome cellulaire. Dans beaucoup de tumeurs, ces points de contrôle sont altérés.
En cas de cancer, les signaux extra-cellulaires ou intracellulaires reçus par la cellule vont être capables d’activer les complexes cycline/cdk ou d’altérer l’activité des inhibiteurs (p21, p15, p16). Le résultat sera la levée du verrou Rb et l’entrée de la cellule en cycle.
Exemple : Le cancer du col de l’utérus : les papillomavirus humains (HPV) sont des petits virus à ADN double brin capables d’infecter les tissus épithéliaux, le plus souvent de façon asymptomatique. Certains types d’HPV dits à haut risque (HPV 16, 18) sont associés au cancer du col de l’utérus. On sait désormais que ce virus s’intègre dans le génome de la cellule hôte où il code pour des protéines virales (e6 et e7) capables de se lier et de dégrader respectivement p53 et Rb, ce qui entraîne une levée du verrou du cycle cellulaire.
Pour plus d’informations, voir le complément en ligne En savoir plus 8.1  : « Rappel sur le cycle cellulaire normal ».
: « Rappel sur le cycle cellulaire normal ».
En savoir plus 8.1 Rappel sur le cycle cellulaire normal
• G1 : post-mitotique. La cellule exerce ses fonctions physiologiques. Durée variable.
• S : duplication de l’ADN durée fixe : 1/3 du cycle.
• G2 : prémitotique, diminution de la synthèse d’ADN, synthèse du fuseau. Durée fixe.
La cellule n’a le choix qu’entre 3 états :
Les régulateurs de cycle cellulaire, en particulier ceux qui contrôlent le passage de la phase G1 à la phase S, sont à l’origine d’évènements oncogéniques dans de nombreux cancers.
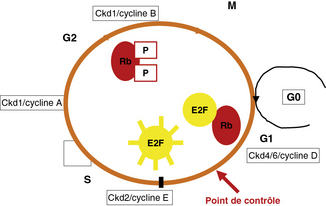
Un des régulateurs essentiels de ce contrôle est la protéine Rb dont le gène est situé en 13q14. Rb est considéré comme le verrou du cycle cellulaire. La forme active (hypophosphorylée) de Rb est capable de bloquer le passage de la phase G0 vers la phase S. La phosphorylation de Rb entraîne la dissociation de Rb du facteur de transcription E2F et l’initiation de la réplication. Cette phosphorylation est sous la dépendance de kinases cycline - dépendantes (cdk) dont l’activation est sous le contrôle de cyclines : couples ckd/cyclines (e.figure 8.1).
Progression tumorale et apoptose
La cellule cancéreuse devient résistante à l’apoptose.
L’apoptose est impliquée dans le contrôle de l’homéostasie cellulaire, et est sous le contrôle de nombreux gènes :
En cas de lésion de l’ADN, le gène P53 est activé, permettant, par l’intermédiaire de p21, l’arrêt du cycle cellulaire et la réparation des lésions de l’ADN ou l’activation de l’apoptose. Il existe des anomalies du gène p53 dans 2/3 des cancers (mutations, délétions) entraînant la suppression du point de vérification de G1 et donc de la voie apoptotique en cas d’instabilité génomique ou d’anomalies chromosomiques.
Dans le lymphome folliculaire, la translocation t(14; 18) aboutit à la juxtaposition du gène BCL-2 avec le locus de la chaîne lourde des immunoglobulines et entraîne la surexpression de la protéine bcl-2. L’accumulation de cette protéine anti-apoptotique augmente la survie des lymphocytes B, ce qui accroît le risque d’acquisition de nouvelles anomalies génétiques conduisant au développement du lymphome folliculaire.
Progression tumorale et immortalité : la cellule cancéreuse a une prolifération illimitée
Les cellules normales sont programmées pour un nombre limité de dédoublements (environ 60–70 in vitro). Aux extrémités des chromosomes se trouvent des séquences répétitives (télomères) qui sont érodées à chaque réplication de l’ADN. Leur disparition induit un arrêt de la prolifération (G0).
Dans la plupart des cellules tumorales, il existe un maintien des télomères au cours des réplications successives. Ceci est dû à la surexpression des télomérases, qui sont les enzymes capables d’ajouter des séquences répétées à l’extrémité des chromosomes.
Modifications fonctionnelles et morphologiques
Fiche signalétique de la cellule cancéreuse
D’un point de vue fonctionnel on reconnaît aux cellules cancéreuses des propriétés communes qui les différencient des cellules normales :
1. indépendance vis-à-vis des signaux de prolifération (facteurs de croissance) provenant de l’environnement ;
2. insensibilité aux signaux anti-prolifératifs ;
4. prolifération illimitée (perte de la sénescence) ;
5. capacité à induire l’angiogénèse ;
6. capacité d’invasion tissulaire et diffusion métastatique.
Ces anomalies fonctionnelles sont l’aboutissement d’un processus multi-étapes dans lequel l’environnement n’est pas neutre. Elles s’accompagnent de modifications morphologiques de la cellule qui permettent le plus souvent de reconnaître son caractère cancéreux en l’observant au microscope optique.
Il faut cependant faire deux remarques :
• aucune de ces anomalies morphologiques prises séparément n’est spécifique de la cellule cancéreuse (en dehors pour certains auteurs des figures de mitoses anormales) ;
• certaines tumeurs au comportement authentiquement malin sont constituées de cellules morphologiquement très proches de leur contrepartie normale ; d’autres critères morphologiques (mauvaise limitation, invasion vasculaire) ou évolutifs (métastases) sont alors nécessaires pour affirmer la malignité.
Modifications du noyau
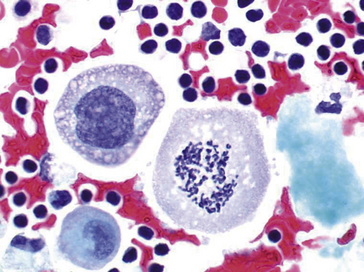
Noyau interphasique
• Anisocaryose (du grec aniso = différent et caryo = noyau) : inégalité de taille d’un noyau à l’autre.
• Augmentation du rapport nucléo-cytoplasmique : le plus souvent due à une augmentation de la taille du noyau.
• Hyperchromatisme : aspect dense et sombre du noyau lié à une condensation ou à une augmentation du nombre des chromosomes (aneuploïdie).
• Irrégularités de forme et de contours (figures 8.5–8.8).
• Multinucléation (figure 8.9).
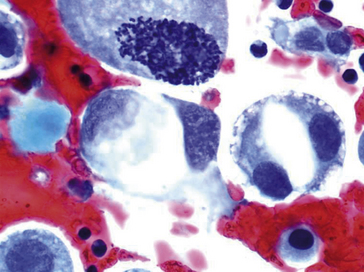
Figure 8.5 Une mitose anormale, noyaux hyperchromatiques irréguliers repoussés par une volumineuse vacuole cytoplasmique. Noter l’anisocaryose : différence de taille des noyaux d’une cellule à l’autre.
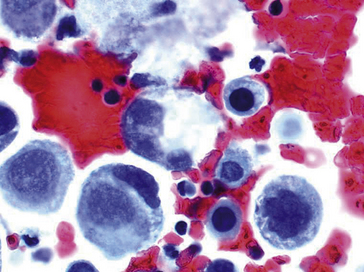
Figure 8.6 Cellules cancéreuses avec noyaux hyperchromatiques et augmentation du rapport nucléo-cytoplasmique. À noter la présence de deux cellules en apoptose.
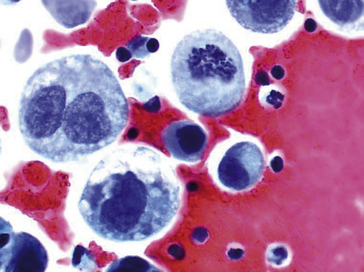
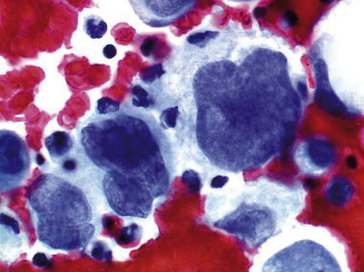
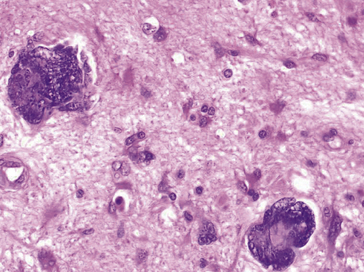
Modifications du cytoplasme
Cytosquelette
Dans la cellule normale, le cytosquelette est constitué de trois types de filaments :
1. microtubules : structures de 20–25 nm d’épaisseur constituées principalement de polymères de tubulines ;
2. microfilaments : structures contractiles de 6–8 nm d’épaisseur contenant notamment des filaments d’actine ;
3. filaments intermédiaires : les plus importants sont les filaments de cytokératine (présents dans les cellules épithéliales et mésothéliales) et de vimentine (surtout dans les cellules conjonctives = mésenchymateuses).
Dans la cellule cancéreuse, le cytosquelette est le plus souvent conservé, avec des anomalies de répartition. Il n’est pas visible en microscopie optique mais ses constituants peuvent être mis en évidence par immunohistochimie. Cette conservation est intéressante pour le pathologiste car la mise en évidence de tels ou tels types de filaments intermédiaires par exemple, permet de préciser le tissu d’origine d’une cellule cancéreuse.
Système sécrétoire
• Variations visibles sur les colorations standards, telles des vacuoles cytoplasmiques (excès de mucus) refoulant le noyau dans les adénocarcinomes mucosécrétants, ou un cytoplasme clair, optiquement vide (accumulation anormale de glycogène) dans les cancers du rein à cellules claires par exemple (figures 8.10, 8.11).
• Variations quantitatives des sécrétions normales (ex : pic d’immunoglobulines monoclonales dans le myélome).
• Apparition de substances nouvelles, soit par dérépression d’une synthèse de protéines de type fœtal (ex : alpha fœtoprotéine, antigène carcino-embryonnaire = ACE), soit par sécrétion inappropriée d’une hormone (ex : sécrétion d’ACTH par certains carcinomes à petites cellules du poumon). Ces substances, considérées comme des marqueurs tumoraux, peuvent être dosées dans le sérum lorsqu’elles sont sécrétées ou identifiées in situ par immuno-histochimie.
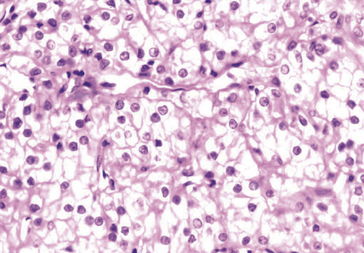
Membrane
La membrane joue un rôle crucial dans les échanges entre les cellules et les interactions avec le milieu extra-cellulaire.
Aspects morphologiques
Les modifications morphologiques ne sont visibles qu’en microscopie électronique : irrégularités, microvillosités, bulles, projections, modifications des systèmes de jonction. Elles ne sont pas prises en compte pour le diagnostic de cancer en routine.
Il existe des modifications des protéines de surface, et notamment des molécules d’adhérence, qui sont impliquées dans les interactions intercellulaires et cellules-matrices extra-cellulaire.
Pour plus d’informations, voir le complément en ligne En savoir plus 8.2 : « Anomalies des molécules d’adhésion ».
En savoir plus 8.2
Anomalies des molécules d’adhésion
Il existe 4 familles de molécules responsables de l’adhésion cellulaire :
• les cadhérines : reconnaissance intercellulaire ;
• des molécules apparentées aux immunoglobulines ;
• les intégrines : reconnaissance de la matrice extra-cellulaire ;
Ces molécules d’adhésion ou CAM (Cell adhesion molecules) ont plusieurs rôles :
Aspects fonctionnels
• Anomalies des récepteurs membranaires : augmentation de nombre et perte de régulation.
• Modifications des enzymes membranaires : augmentation des enzymes protéolytiques (protéases, glycosidases) favorisant la dégradation de la substance intercellulaire.
• Modifications des antigènes de membrane :
Stroma tumoral
Le stroma tumoral est caractérisé par tout ce qui est présent au sein d’une tumeur et n’est pas une cellule tumorale. Le stroma comprend donc le tissu conjonctif, les vaisseaux, les leucocytes et la matrice extra-cellulaire.
Le stroma sert de charpente à la tumeur et assure ses apports nutritifs. Il est sous la dépendance du tissu tumoral dont les cellules peuvent, par exemple, élaborer des substances qui vont favoriser la pousse des vaisseaux. Il est d’usage de réserver le terme de stroma au support conjonctif des tumeurs malignes et de ne pratiquement pas l’utiliser dans le cas des tumeurs bénignes, mais rien ne s’y opposerait conceptuellement.
C’est dans les carcinomes invasifs que le stroma est le plus nettement individualisé. Il y a cependant un stroma dans toutes les autres tumeurs solides, constitué au minimum des vaisseaux et d’une matrice extra-cellulaire d’abondance variable.
Les variations morphologiques du stroma sont multiples, certaines d’entre elles sont caractéristiques d’un type tumoral donné et auront donc une valeur séméiologique pour le diagnostic du type tumoral (figure 8.12).
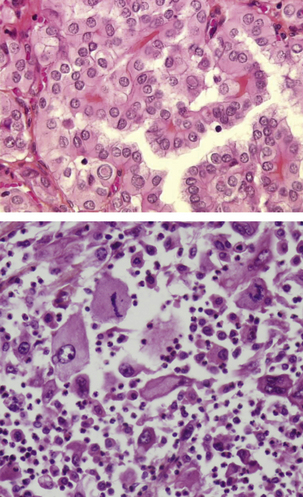
Figure 8.12 Stroma tumoral : dans ces 2 carcinomes thyroïdiens, le stroma est pratiquement absent, réduit à quelques capillaires, dans une variante bien différentiée (en haut), ou particulièrement abondant et riche en polynucléaires dans une variante anaplasique (en bas).
Variations quantitatives
Certains carcinomes très différenciés ont un stroma qui peut être exactement proportionné à la prolifération épithéliale. Dans les tumeurs endocriniennes, le stroma comporte souvent des capillaires sinusoïdes semblables à ceux d’une glande endocrine normale (stroma adaptatif).
Plus souvent, le stroma est disproportionné par rapport à la prolifération épithéliale :
• lorsqu’il est relativement peu abondant, la tumeur sera molle, souvent nécrosée, semblable macroscopiquement à du tissu cérébral. C’est un cancer que l’on caractérisera macroscopiquement d’« encéphaloïde » ;
• à l’inverse, lorsqu’il est très abondant, riche en fibres collagènes, la tumeur sera dure et rétractée, c’est le squirrhe. Cette rétraction, comparable à celle de certaines cicatrices pathologiques, est liée à la présence de nombreux myofibroblastes.
Variations qualitatives
Le tissu conjonctif du stroma possède certaines propriétés réactionnelles du tissu conjonctif normal. Il peut s’y produire une réaction inflammatoire. Celle-ci surviendra, par exemple, lors de la destruction du tissu tumoral par une irradiation. La nécrose des cellules tumorales déclenche une réaction exsudative. Il peut même se produire une réaction à corps étrangers autour de squames de kératine élaborées par la tumeur. Dans certaines tumeurs, la réaction inflammatoire du stroma est une réaction tuberculoïde.
Quelques tumeurs ont un stroma riche en cellules lymphocytaires ou plasmocytaires, ce qui peut être la manifestation d’une réaction immunitaire. Cet aspect va parfois de pair avec un pronostic meilleur.
Cancer et angiogenèse
La néovascularisation issue de l’angiogenèse tumorale présente un état d’activation cellulaire maximum pour une efficacité de perfusion médiocre. Elle est très hétérogène en densité, par sa maturation phénotypique d’une zone tumorale à l’autre et d’une tumeur à l’autre.
Une tumeur ne peut pas croître au-delà de 1 à 2 mm sans l’aide d’une riche vascularisation sanguine. Les rapports entre le tissu tumoral proprement dit et sa vascularisation sont donc critiques dans l’histoire naturelle de chaque cancer.
La vasculogenèse est une prolifération vasculaire due à la différenciation de cellules précurseurs, communes aux lignées sanguines, en cellules endothéliales qui se répandent, s’associent et établissent un réseau vasculaire. Ce terme est très majoritairement réservé aux étapes correspondantes de l’embryogenèse.
L’angiogenèse est une prolifération vasculaire due au bourgeonnement vasculaire à partir de vaisseaux préexistants, puis à l’installation d’un réseau et à sa différenciation en différents secteurs fonctionnels. Ce processus implique le recrutement et la différenciation de cellules péricytaires et de cellules musculaires lisses, qui concourent à stabiliser le nouveau réseau et à lui donner une efficacité fonctionnelle. L’angiogenèse est souvent liée aux processus inflammatoires ou tumoraux.
Vascularisation en périphérie des tumeurs
Dans la zone périphérique d’invasion tumorale, la prolifération des cellules endothéliales est active et elle produit de nouveaux vaisseaux souvent anormaux. La prolifération vasculaire est particulièrement vigoureuse et l’index de prolifération des cellules endothéliales est 50 à 200 fois plus élevé que pour les mêmes cellules des tissus normaux.
Les vaisseaux créés au sein de la tumeur sont anormaux. Ce sont des canaux à paroi mince plutôt de type veinulaire, irrégulièrement anastomosés avec de nombreux culs-de-sac. Ils ont tendance à former des shunts artério-veineux. La bordure endothéliale est incomplète (sauf dans les tumeurs cérébrales primitives), la membrane basale est souvent absente, les cellules satellites (péricytes et cellules musculaires lisses) raréfiées. Il n’y a pas d’innervation et de nombreux espaces vasculaires sont bordés directement par les cellules tumorales.
Ces vaisseaux défectifs ne sont pas contrôlables par les mécanismes locaux habituels (mécanisme nerveux et système des cytokines). L’efficacité de perfusion est médiocre. Les courts-circuits artério-veineux s’opposent à une perfusion capillaire efficace. Le régime liquidien est chaotique avec des inversions de flux et une stase selon une période de 2–3 min.
Le drainage des fluides interstitiels est déficient en liaison avec l’excès de perméabilité et l’absence de drainage lymphatique fonctionnel.
Enfin, cette vascularisation est très inégalement répartie en densité d’un point à un autre de la tumeur.
Dans cette région de la tumeur on retrouve des taux élevés de facteur de croissance endothélial vasculaire (VEGF), du facteur de croissance fibroblastique basique (FGFb), de la phosphorylase de la thymidine (TP). Tous ces facteurs sont induits par l’hypoxie.
Vascularisation au centre des tumeurs
Au fil de la croissance tumorale, les marges s’incorporent dans le centre de la tumeur, mêlant néovascularisation et vascularisation d’origine de l’hôte. La densité de microcirculation devient 4 à 10 fois plus faible qu’au niveau des berges. Les cellules tumorales s’adaptent à l’hypoxie en activant la glycolyse anaérobie. Les cellules endothéliales activent la fabrication des molécules du stress hypoxique (VEGF, TP, complexe VEGF/récepteur du VEGF) et les inhibiteurs de l’apoptose (bcl-2). Quand le mécanisme anti-apoptotique endothélial défaille, les cellules tumorales sont en situation d’accès facile au compartiment intravasculaire.
Immunité anti-tumorale
La réponse immune joue un rôle majeur dans la défense de l’organisme contre les tumeurs, et est probablement responsable du contrôle et de la majorité des tumeurs. Ceci est notamment valable à la phase initiale d’émergence des tumeurs, mais l’infiltration tumorale par des lymphocytes à un stade plus évolué reste un facteur pronostic important pour plusieurs tumeurs.
Effecteurs de la réponse immune anti-tumorale
La réponse immune anti-tumorale fait intervenir :
• l’immunité innée, avec notamment des cellules cytotoxiques (ex : lymphocytes NK), et des facteurs solubles (ex : interféron gamma), qui peuvent avoir des effets directs ou indirects (pro-inflammatoire ou anti-angiogénique) ;
• l’immunité adaptative, c’est-à-dire dépendante de la reconnaissance de molécules spécifiques produites par la tumeur.
Les mécanismes effecteurs de la réponse immune anti-tumorale sont :
• la cytotoxicité directe par les lymphocytes NK (NK = natural killer), les lymphocytes T cytotoxiques (CD8), ou les cellules dendritiques IKDC (Interferon gamma producing killer dendritic cells) (tableau 8.4) ;
• la cytotoxicité médiée par les anticorps, qui paraît notamment très utile en thérapeutique, avec l’utilisation d’Ac monoclonaux spécifiques de certains antigènes exprimés par les tumeurs (CD20, EGFR) ;
• la production de facteurs solubles capables de moduler la réponse inflammatoire locale et/ou l’angiogénèse, tels l’interféron gamma.
Tableau 8.4 Mécanismes effecteurs de la réponse immunitaire anti-tumorale par cytotoxicité directe.
| Cellule effectrice | Ligand sur la tumeur | Apoptose induite par |
| Lymphocyte T CD8 | Restreint au CMH1 | perforine |
| Lymphocyte NK | Non restreint au CMH1 Expression par la tumeur de MICA ou MICB qui active NKG2D sur les NK |
perforine ou TRAIL* |
| Cellules dendritiques IKDC | perforine ou TRAIL* |
* Tumor necrosis factor receptor related apoptosis inducing ligand
Échappement des tumeurs à la réponse immune
Les mécanismes d’échappement des tumeurs concernent à la fois la réponse immune innée et adaptative. Il peut s’agir :
• d’une immuno-sélection : sélection au cours du temps des sous-clones tumoraux ayant acquis des mécanismes d’échappement à la réponse immune. Ces sous-clones sont généralement sélectionnés en raison de la diminution de l’expression de cibles ou l’augmentation de l’expression d’inhibiteurs ;
• d’une immuno-subversion (induction d’une tolérance spécifique) mettant en jeu des phénomènes plus complexes de coopération intercellulaire.
Pour plus d’informations, voir le complément en ligne En savoir plus 8.3 : « Exemples de mécanismes impliqués dans l’immuno-sélection ».
En savoir plus 8.3
Exemples de mécanismes impliqués dans l’immuno-sélection
Diminution de l’expression de :
• complexe majeur d’histocompatibilité de classe I (CMH1) et/ou des transporteurs de peptides (TAP1), qui sont nécessaires pour l’activation des lymphocytes T CD8;
• MICA, qui est un ligand du récepteur NKG2D activant les lymphocytes NK ;
• récepteurs de mort Fas, TNFR1 et TRAIL1 et TRAIL2, ou de la caspase 8 qui sont des voies effectrices de l’apoptose des cellules tumorales.
Stratégies thérapeutiques immunologiques
Pour les tumeurs viro-induites la stratégie vaccinale peut être efficace. Ainsi la vaccination d’une population contre l’hépatite B permet de prévenir la survenue d’hépatites chroniques B et de réduire de façon importante l’incidence du carcinome hépatocellulaire qu’elle aurait induit.
L’immunothérapie par instillation du vaccin BCG en intravésical est utilisée depuis de nombreuses années pour contrôler l’évolution des tumeurs superficielles de vessie de haut grade.
Des injections d’interleukine 2 peuvent induire des régressions métastatiques dans certains cancers du rein ou dans les mélanomes.
Actuellement, plusieurs anticorps monoclonaux sont dirigés contre des antigènes spécifiques des tumeurs. Ces Ac sont éventuellement associés à des radio-éléments ou des toxines.
Pour plus d’informations, voir le complément en ligne En savoir plus 8.4 : « Exemples d’anticorps monoclonaux anti-tumoraux ».
En savoir plus 8.4
Exemples d’anticorps monoclonaux antitumoraux
• L’anticorps monoclonal Trastuzumab (Herceptin) est dirigé contre un des corécepteurs de la famille des récepteurs de type I des facteurs de croissance, la protéine HER2. La détection sur coupe histologique de la surexpression de la protéine ou de l’amplification du gène correspondant dans les cellules tumorales de carcinome du sein est retrouvée dans 15 % des patientes et rend ces dernières éligibles pour le traitement.
• D’autres anticorps monoclonaux bénéficient aussi actuellement d’une AMM, tels les anti-CD20 dans des lymphomes CD20 + et leucémies B, et les anti-récepteur de l’EGF dans des carcinomes colorectaux avancés.
Ces anticorps, utilisés comme traitements anti-tumoraux, posent de redoutables problèmes techniques, donc économiques. Ces anticorps sont au départ murins, ce qui induit leur neutralisation dans les deux semaines suivant le début du traitement, temps requis pour l’induction d’une réponse anti souris à anticorps de haute affinité chez le patient. Il y a donc nécessité d’« humaniser » ces anticorps, ce qui en fait actuellement des médicaments extrêmement coûteux. Ces stratégies thérapeutiques sont parallèles mais non identiques à celles qui visent à inhiber la fonction des récepteurs membranaires des cellules tumorales par des drogues.
 L’essentiel à retenir
L’essentiel à retenir La très grande majorité des cancers résulte des effets cumulés d’altérations survenues successivement sur l’ADN des cellules tumorales. Ces altérations sont responsables de la prolifération et l’accumulation de cellules d’origine monoclonale ou oligoclonale, puis du développement de sous-clones.
Les trois principales familles de gènes impliqués dans la cancérogenèse sont les oncogènes, les gènes suppresseurs de tumeur et les gènes responsables du maintien de l’intégrité du matériel génétique.
Les altérations responsables de la dérégulation de l’expression ou de la fonction de ces trois grandes familles de gènes peuvent être des anomalies chromosomiques, génétiques ou épigénétiques.
Les oncogènes et gènes suppresseurs de tumeur sont le plus souvent impliqués dans la régulation des grandes fonctions cellulaires, et notamment le cycle cellulaire, l’apoptose et la signalisation intracellulaire.
Les facteurs de risque de développement des cancers sont héréditaires (monogéniques ou familiaux) ou environnementaux (virus, radiation, toxique).
Les interactions entre la tumeur et son hôte (le patient), en particulier l’angiogenèse et la réponse immune, constituent des modes de régulation majeurs de la croissance tumorale.
Pour les entraînements du chapitre 8, cf. p.182.